
Introduction
In December 2019, in the Chinese city of Wuhan, there have been reports of an outburst of clinical cases of patients with pneumonia-like symptoms as a result of an infection caused by a novel virus belonging to the coronavirus family.1 Molecular characterization as per World Health Organization (WHO) standards, categorized it as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV2), based on which it was further named as novel coronavirus-2019 (2019-nCoV), and hence the disease was named as coronavirus disease-2019 (COVID-19).2 It has been recognized as a highly contagious and pathogenic virus that may cause a host of symptoms ranging from mild cold to fatal illnesses triggered by acute respiratory distress syndrome (ARDS). Cytokine storm, an important causal factor of ARDS, results in excessive and uncontrolled release of pro-inflammatory cytokines. Recent reports suggest that elevated cytokine profile that includes interleukin-1 (IL-1), interleukin-2 (IL-2), interleukin-10 (IL-10) and tumour necrosis factor-alpha (TNFα) are abnormally elevated in a COVID-19 patient’s blood serum.3 Cytokine storm results in abnormally high cell inflammation that leads to acute lung injury, thus resulting in ARDS, in addition to extrapulmonary systemic hyperinflammation syndrome. Hence, SARS-CoV2 infection causes cytokine storm that in turn triggers ARDS and multiple organ failure, resulting in mortality.4 In order to minimize the severity of SARS-CoV2 infection, attenuation of the cytokine storm may be an essential step. Among the recently reported methods, low-dose corticosteroids treatment inhibits secretion of pro-inflammatory markers including IL-1, IL-6, granulocyte colony stimulating factor (GCSF), TNF-α and cyclooxygenase (COX2) synthesis, thus minimizing severe lungs damage associated with COVID-19.5 However, inflammation inhibition alone cannot overcome COVID-19. Identification of therapeutic molecule that has dual properties of anti-inflammation and anti-viral multiplication may lead to an ideal drug treatment against COVID-19.
Figure 1
Chemical structure of the following compounds (A) Brassinolide; (B) 28-Homocastasterone; (C) 24-epibrassinolide (24-EB); and (D) Hydroxychloroquine (HCQ)
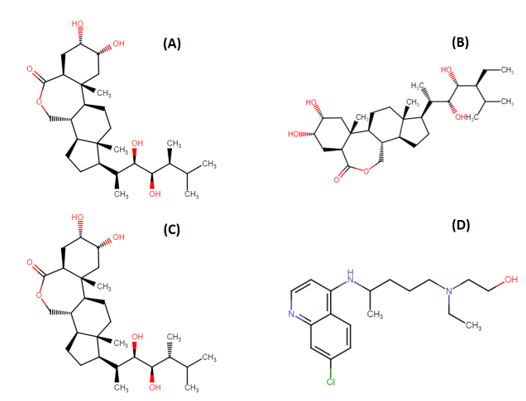
Brassinolide (BS), 28-Homocastasterone (28-HC) and 24-epibrassinolide (24-EB) are plant hormones belonging to brassinosteroid family (Figure 1). They are biosynthesized by CYP72B1 enzyme and regulates a wide range of physiological and metabolic processes including seed germination, senescence and stress response.6 Previous studies suggest that phytosteroids have antiviral potential against herpes simplex virus, Junin virus, arena viruses, and vesicular stomatitis viruses.7, 8 Mechanism of action of these phytosteroids mainly focuses on inhibition of viral multiplication.9 Conventional pharmacological methods of drug discovery are time-consuming, labour-intensive and expensive. An alternative concept of reverse pharmacology could be a major breakthrough in the field of drug discovery. The basis of reverse pharmacology includes in-silico analogue designing and ligand-receptor interaction.10
Figure 2
PDB structures of the following SARS-CoV proteins: (A) Spike protein; (B) Nucleocapsid protein; (C) Main protease
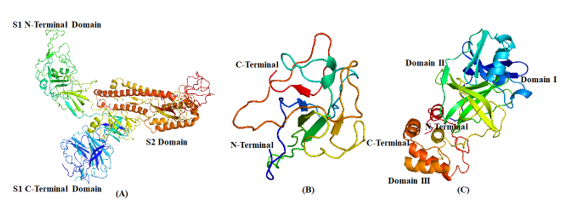
In the present in-silico study, we intend to investigate the anti-viral potential of specific phytosteroids, namely, BS, 28-HC and 24-EB, against SARS-CoV2 protein and human pro-inflammatory cytokines as an ideal drug candidate. These potential phytosteroids are hypothesized to have the ability to ct as inhibitor of viral proteins including Spike-protein (S-protein) subunit 1 (S1), nucleocapsid protein (N-protein), and main protease (Mpro) (Figure 2), in addition to the host pro-inflammatory proteins COX-2, prostaglandin synthase (PS), IL-1 and TNF-α. The docking studies was performed by using hydroxychloroquine (HCQ) as standard drug against the phytosteroids.
Materials and Methods
Compound preparation
In-silico structures of four molecules, namely, BS (CID:115196), 28-HC (CID:11038340), 24-EB (CID:443055) and HCQ (CID:3652) were downloaded from PubChem database (http://pubchem.ncbi.nlm.nih.gov/) as .SDF file format. The files were converted to .mol2 format using Open Babel software version 2.4.1. and optimized by means of ligand preparation script in AutoDock ver. 4.0. program. The inhibitors were prepared for docking as detecting root, torsion tree were set and saved in .pdbqt file format.
Protein structure and preparation
3D structures of S-protein S1 subunit (PDB ID: 6acc), N-protein (PDB ID: 4kxj) and Mpro (PDB ID: 6lu7), COX-2 (PDB ID: 4m1), PS (PDB ID: 1z9h), IL-1(PDB ID: 2nvh) and TNF-α (PDB ID: 2az5) were retrieved from RCSB protein databank (http://www.rcsb.org). AutoDock Tools (http://autodock.scripps.edu/resources/adt) facility supported the protein set up for docking. The script procedure employs the following ADT routines including removal of water molecules, addition of polar hydrogen atoms, assignment of Kollman charges and conversion of the protein files in. pdb format for molecular docking.
Grid box generation
3D structures of selected proteins and chemical structures were complexed together to forrm a grid. Therefore, the centroid of the small molecule in complex structures was chosen to generate grid points X = 60, Y = 60 and Z = 60 axis set for docking analysis. The grid file was generated by means of “grid generation panel” in AutoDock software ver. 4.0.11
Molecular docking simulation
Protein-ligand docking simulations were performed using AutoDock4 tool. For each ligand (chemical structure), 100 docking runs with default parameters were performed by treating protein as rigid and the ligand as flexible. The results were visualized using PyMol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrodinger, LLC.), wherein all the conformations for each of the ligand was found to be within the cavity of protein indicating that the docking run was free from errors. The conformational clusters with lowest binding energy were considered for further analysis.10
Evaluation of the total binding energy
The AutoDock ver. 4.0 algorithm was applied to evaluate the total binding energy of phytosteroids against various proteins. Various docked conformations were obtained and the ones with lowest binding energy towards ligand binding cavity of protein were selected as the possible binding conformation, and considered for further interaction analysis. The final evaluations of the interactions between the target compounds and amino acid residues of the ligand binding cavity of protein were analyzed using online Protein Ligand Interaction Profiler tool (https://plip.biotec.tu-dresden.de/plip-web/plip/index).
Results
The SARS-CoV2 S1 subunit (S1) binding interaction with dietary phytosteroids were studied by in-silico docking simulations. Based on docking energies obtained, S1 exhibited highest binding affinity towards BS, followed by 28-HC, and 24-EB, thereby exhibiting -7.24, -8.27 and -8.2 Kcal/mole respectively, as compared to HCQ that exhibited binding affinity of -6.36 Kcal/mole. In S1-BS interaction, BS interacted with LYS929 residues via hydrogen bond and PRO710, ASP932, VAL 933, GLU999, ILE1000, LEU1006 residues via hydrophobic interaction (Table 1 and Figure 3). Similarly, in S1-28-HC interaction, GLY981 and GLN984 amino acid residues form hydrogen bonds with 28-HC, while THR943, LEU944, GLN947, GLN984, THR988 and LEU1006 residues form hydrophobic interactions (Table 1). Likewise, the binding studies performed between 24-EB and S1, indicates 24-EB interacting with amino acid residues GLY981, GLN984, SER985 via hydrogen bond, and with amino acid residues THR943, LEU944, GLN947, GLN984, THR988, TYR989 via hydrophobic interaction (Table 1 and Figure 3). HCQ, on the other hand, interacts with S1 amino acid residues GLN992 and ARG996 via hydrogen bond, while hydrophobic interactions are observed with ALA940, THR943, THR988, TYR989 amino acid residues with lowest binding affinity (-5.22 Kcal/mole) as compared to the three dietary phytosteroids (Table 1 and Figure 3).
Table 1
Molecular docking analysis ofSpike (S) protein S1 subunit interactions with various ligands
Table 2
Molecular docking analysis ofnucleocapsid protein interactions with various ligands
Table 3
Molecular docking analysis ofmain protease interactions with various ligands
Table 4
Molecular docking analysis ofinterleukin-1 cytokine interactions with various ligands
Table 5
Molecular docking analysis oftumor necrosis factor-α cytokine interactions with various ligands
Table 6
Molecular docking analysis ofcyclooxygenase-2 interactions with various ligands
Table 7
Molecular docking analysis ofprostaglandin synthase interactions with various ligands
In the current in-silico study, N-terminal domain (NTD) were docked with the dietary phytosterols and HCQ. The docking scores of BS, 28-HC, and 24-EB against NTD of N-protein were -11.24 Kcal/mole, -10.80 Kcal/mole and -11.23 Kcal/mole, respectively compared to HCQ, that scored -7.16 Kcal/mole of binding energy with NTD of SARS-CoV2 (Table 2 and Figure 4). The BS forms hydrogen bond with VAL59, SER64 and ARG164 amino acid residues, while it forms hydrophobic interaction with PHE57, PRO61, HIS104, and TYR124 amino acid residues of NTD of N-protein. Likewise, 28-HC forms hydrogen bond with SER64 and TYR102 residues, while it forms hydrophobic interaction with PHE57, PRO61, TYR102, HIS104, and TYR124 amino acid residues of NTD of N-protein (Table 2). Both BS and 28-HC ligands form π-stacking interaction with TYR63, TYR124 amino acids residues. Correspondingly, 24-EB forms hydrogen bond with TYR62, SER64, and TYR102 amino acid residues and hydrophobic interaction with PHE57, VAL59, PRO61, TYR63, TYR102, HIS104, and TYR124 residues of NTD of N-protein (Table 2 and Figure 4). Hence, π-stacking interaction of N-protein’s NTD with TYR63 and TYR124 amino acids residues is common to all the three phytosterols. Docking studies between HCQ ligand and NTD of N-protein revealed hydrogen bonding between HCQ and amino acid residues ASN58 and VAL59, while PHE57, PRO61, TYR102, TYR124 are involved in hydrophobic interaction with HCQ.
Figure 3
The molecular docking of SARS-CoV-2 Spike (S) protein S1 subunit interaction with ligands. (A, C, E, G) The putative binding site of BS and 24-BC 24-Epi and HOC on SARS-CoV2 protein 3-D ribbon structure of SARS-CoV2 protein. (B, D, F, H). The interacting amino acid residues of SARS-CoV-2 with BS, 24-BC, 24-Epi and HOC.
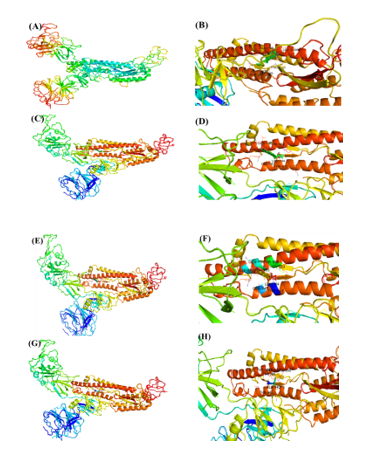
Figure 4
The molecular docking of SARS-CoV2 Nucleocapsid protein interaction with ligands. (A, C, E, G) The putative binding site of BS and 24-BC 24-Epi and HOC on SARS-CoV2 protein 3-D ribbon structure of SARS-CoV2 protein. (B, D, F, H). The interacting amino acid residues of SARS-CoV-2 with BS, 24-BC, 24-Epi and HOC.
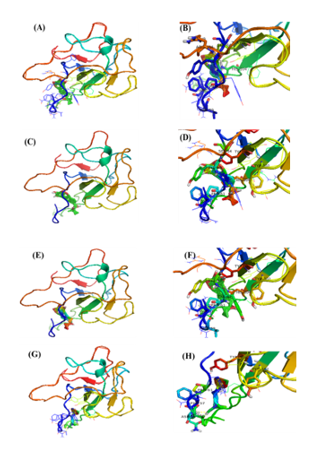
Figure 5
The molecular docking of SARS-CoV2 Main protease protein interaction with ligands. (A, C, E, G) The putative binding site of BS and 24-BC 24-Epi and HOC on SARS-CoV2 protein 3-D ribbon structure of SARS-CoV2 protein. (B, D, F, H). The interacting amino acid residues of SARS-CoV-2 with BS, 24-BC, 24-Epi and HOC.
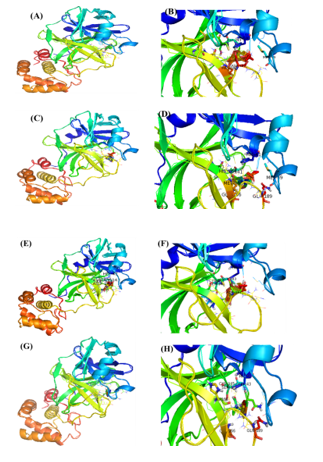
Docking scores of BS, 28-HC, and 24-EB against Mpro were -10.36, -9.25, and -10.16 Kcal/mole respectively (Table 3 and Figure 5), whereas HCQ scored -7.90 Kcal/mole of binding energy against Mpro. Among the three phytosterols, BS had the highest docking score and consequently highest binding affinity for Mpro, thereby forming hydrogen bond with GLY 143, SER144, CYS145 and HIS163 residues, while HIS41, MET49, MET165 and ASP187 residues were involved in hydrophobic interaction. It also formed π-Stacking bond formed with HIS41 amino acid of Mpro (Table 3 and Figure 5). In comparison to BS, HCQ formed hydrogen bonds with PHE140, GLY143, SER144, CYS145 and HIS163 residues, while HIS41, MET49, MET165 and ASP187 of Mpro formed hydrophobic interaction.
Docking scores of BS, 28-HC and 24-EB against pro-inflammatory protein, IL1, are -9.02 Kcal/mole, -13.83 Kcal/mole, and -9.97 Kcal/mole, respectively (Table 4). Whereas, the same series of phytosterols has a docking score of -9.96 Kcal/mole, -6.07 Kcal/mole, and -5.51 Kcal/mole respectively against TNF-α (Table 5). Docking scores of phytosterols against pro-inflammatory COX-2 are -12.25 Kcal/mole, -13.77 Kcal/mole, and -13.22 Kcal/mole (Table 6), whereas the same series of phytosterols when docked against PS are -8.01 Kcal/mole, -8.95 Kcal/mole, and -9.48 Kcal/mole (Table 7). Similarly, HCQ’s docking score against IL1 and TNF-α are -4.99 Kcal/mole and -7.51 Kcal/mole respectively (Table 4, Table 5). Consequently, HCQ’s docking score against pro-inflammatory enzymes, COX-2 and PS are -10.44 Kcal/mole and -6.52 Kcal/mole (Table 6, Table 7) respectively.
The common molecular interaction between BS, 28-HC, and 24-EB against IL1 and TNF-α amino acid residues are (LYS77, LEU80, PHE133, LEU134) and (LYS65, THR77, TYR115, ASP143, ALA145, GLU146, GLN149) respectively whereas HCQ does not show significant similarity of molecular interaction with two proteins. Similarly, common molecular interaction between BS, 28-HC, 24-EB against the two targets, COX2 and PS, include amino acid residues (ALA199, PHE200, ALA202, PHE210, TYR385, TRP387, LEU390, LEU391) and (PHE112, VAL148, ASP164, SER165, LYS208, TYR209) respectively.
Discussion
Phytosteroids are consumed as part of functional food and herb-based folk medicine. 12 Assimilation of phytosteroids into vital organs via blood circulation induces metabolic changes in mammalian cells that includes antihyperglycemic and alterations of pro-inflammatory cytokines including IL-1, TNF- α, and COX-2. 13, 14 Three-dimensional structure of IL-1 consists of open-ended barrel shape composed beta pleated strands with two IL-1 receptor binding sites. 15 3D-structure of TNF-α consists of three monomers to form a compact bell-shaped trimer. TNF-α monomer forms an elongated, antiparallel beta-pleated sheet with a jelly-roll topology. 16 COX2 converts arachidonate into prostaglandins and thromboxane, thus, contributing to acute and chronic inflammatory modulatory effects. 17
Structurally, SARS-CoV2 is a nanometre-sized entity ranging between 65-125 nm in diameter. It has non-segmented positive direction RNA of 26-32kbs in length with crown-shaped spikes on the outer surface. It contains four structural proteins, namely, S-protein, E-protein, M-protein, and N-protein. 18 S-protein is a type I transmembrane glycoprotein of approx. 150 kDa molecular weight and is composed of two subunits, namely, S1 and S2, protruding from the viral surface. The S1 subunit (Figure 2 a) binds to angiotensin 2 converting enzyme (ACE2) host cellular receptor through its receptor-binding domain (RBD), followed by conformational changes in the S2 subunit, which allows the fusion peptide to insert into the host cell membrane. 19 Similarly, N-protein (Figure 2 b), a major structural protein that plays various roles in the viral replication and forms a ribonucleoprotein complex with the viral RNA through its NTD. The SARS-CoV2 main protease (SARS-CoV2 Mpro) consist of three domains that processes polypeptide with the help of a catalytic dyad and the active site of the protease is located between the domains I and II (Figure 2 c). 20 The spikes are responsible for the attachment of the viruses on the host surface and their subsequent entry into the host cells. Similarly, the N-protein and main protease (Mpro) participates in packaging and multiplication of the virus. Disruption of virus entry into the host cells and virus replication machinery may be potential therapeutic targets. Development of S-protein, N-protein and Mpro-targeted drugs against coronaviruses may be probable therapeutic option. 21 So far, no specific drug against COVID 19 has been approved by the Food and Drug Administration Agency (USFDA). 22 Initiation of SARS-CoV2 infection requires successful penetration of the virus in host cell membrane barrier followed by modulation host cellular metabolic stimuluses that may recognise the virus via a number of physicochemical and cell-signalling methods. Host cellular modulation aids in virus multiplication by influencing the homeostatic regulatory processes that controls cell and tissue functions. It ensures equilibrium dynamics of factors such as rate limiting steps, circulatory partitioning, product inhibition, protein synthesis and gene expression that affects respiratory, hepatic, gastrointestinal and neurologic homeostatic processes.13
Natural compounds from plant sources are among the potential candidates that are currently being investigated for their influence on viral cellular metabolic processes, thus acquiring early translational knowledge of such compounds as potential therapeutic interventions against COVID-19. The current study was undertaken to identify probable phytochemical candidates that could bind to the spike S1 subunit of S-protein, N-protein and Mpro, all of which are understood to be potential drug-targets against SARS-CoV2. Among the various phytochemicals tested against these three protein targets, we identified three dietary phytosteroid molecules that could bind with the inhibitor binding domain of the target proteins, as compared to an anti-malarial drug, HCQ, which is currently being repurposed for managing COVID-19, due to lack of any standard drug. 23 HCQ as the choice of positive control standard drug may be debatable. However, our choice of HCQ was based on recent evidences of COVID-19 response to HCQ treatment. One of the recent reports published in The Lancet Respiratory Medicine Journal suggest that in absence of any significant alternative, clinical trials are being conducted on HCQ with some significant results, especially in critically ill COVID-19 patients with history of pneumonia. 24 Another report of experiments conducted on COVID-19 infected non-human primates suggest that the cytokine storm was significantly attenuated by the HCQ treatment. 25 The molecular mechanism of action of HCQ on inhibition of cytokine storm in non-COVID-19 patients, however, is well established, especially in rheumatoid arthritis and Systemic lupus erythematosus. 26, 27
Comparatively low binding affinity of HCQ with S1 may be due to sterically bulkier carbon chain which could disrupt the interaction of HCQ with the binding site, thus contributing to the significantly lower binding affinity. 28 Both phytosteroids, 28-HC and 24-EB, forms hydrogen bond with GLY981 and GLN984 of S1 subunit, while they both form of hydrophobic interactions with amino acid residues THR943, LEU944, GLN947, GLN984, THR988 of the S1 subunit protein. HCQ forms lesser number of hydrophobic interaction and hydrogen bonds with the amino acid residues of S1 subunit, thus, further suggesting the reason behind low binding affinity of HCQ in comparison to phytosteroids, 28-HC and 24-EB.
N-protein binds to the viral RNA genome, forming a helical ribonucleoprotein (RNP) or nucleocapsid structure complex. RNP maintains RNA conformation that is necessary for replication and transcription of viral genome, in addition to cell-cycle progression via actin reorganization. N-protein consists of three domains, namely, N-terminal RNA-binding domain (NTD), C-terminal dimerization domain (CTD) and a central Ser/Arg (SR)-rich linker region. 21 Previous studies suggests that the N- and C-terminal domains of the N proteins are responsible for RNA binding and oligomerization, respectively. 28
Mpro, a 306 amino acids enzyme, induces the spike protein that further helps in viral penetration. 29 The current study was undertaken to identify phytosterols as a potential drug candidate against COVID-19 that could significantly bind and inhibit Mpro enzymes. Except for 28-HC, all other ligands formed π-Stacking interaction with HIS-41 amino acid residue, indicating that the amino acid HIS-41 may play an important role in inhibitor-protein binding affinity, thus, displaying lower binding energy.
Figure 6
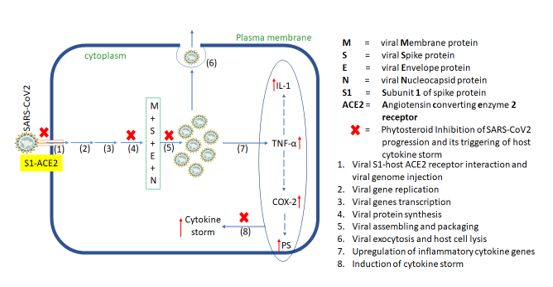
Schematic illustration of the proposed mechanism of action of phytosteroids against COVID-19 prophylaxis. Phytosteroids may have multiple levels of mechanism of action. They may prevent viral entry into the host by preventing binding of viral subunit-S1 with ACE2 receptor. mechanism in inhibition of virus’ host entry and multiplication. If the virus is able to bypass this inhibition, these phytosteroids may prevent viral multiplication by inhibiting protein synthesis and vial components assembling. In case, the second level of defence also fails, the virus may trigger upregulation and activity of pro-inflammatory cytokines resulting in cytokine storm. Phytosteroids may attenuate this cytokine storm as the last resort to overcome manifestations of SARS-CoV2 infection.
IL-1 IL-6, GCSF and TNF-α are some of the pro-inflammatory cytokines produced by activated macrophages. Low-dose corticosteroids treatment lowers expression levels of these pro-inflammatory markers, in addition to quenching of COX-2 synthesis. These biomarkers are otherwise abnormally elevated as part of the natural immune response and may have a role in severe lung damage associated with COVID-19. 30 Therefore, to overcome these acute and chronic inflammatory modulation effects, in-silico molecular docking studies of potential anti-inflammatory phytosteroids were undertaken to identify potential alternative therapeutic molecules against COVID-19. The docking scores of BS, 28-HC, 24-EB and HCQ against two pro-inflammatory markers and two pro-inflammatory enzyme targets have been compared. BS has the highest docking score against all four pro-inflammatory targets suggesting that BS could have superior anti-inflammatory activity as compared to other two phytosteroids and HCQ.
Inhibition of host cell entry and viral replication machinery may be ideal therapeutic targets, but a parallel defence mechanism via controlling of cytokine storm gives therapeutic edge at two levels against SARS-CoV2. In case, a virus escapes firstly line of defence and is able to replicate inside host cell, a second level of defence may be triggered which attenuates cytokine storm from getting triggered. Hence, we identified seven such target proteins, S-, N-, and Mpro proteins, IL-1, TNF-α, COX2 and PS that may be potentially targeted by these three phytosteroids. Hence, based on the above hypothesis and its corresponding in silico results, we proposed a plausible molecular mechanism of action of these phytosteroids wherein viral entry points and multiplication may be hindered. If at all the virus succeeds in multiplication and trigger cytokine storm, these steroids may counter inhibit the expressions and activity of the corresponding cytokines (Figure 6). In summary, SARS-CoV2-related proteins are involved in host cell entry and viral replication, while the cytokine proteins are involved in triggering cytokine storm. All the seven target proteins were docked against the three phytosterols, BS, 28-HC, and 24-EB in comparison to the current repurposed drug, HCQ, wherein the docking scores and in turn the binding affinity of BS is highest, thus, suggesting that BS may be an effective alternative to HCQ against COVID-19.
Conclusions
The present in-silico study suggests that BS is an effective alternative to HCQ since it may potentially inhibit COVID-19 at multiple levels including host entry, viral multiplication and attenuation of cytokine storm. The outcome of our in-silico data may be the basis for in vivo and in vitro studies against COVID-19, simultaneously, establishing the concept of reverse pharmacology.